
Le laboratoire Matériaux et Phénomènes Quantiques (MPQ) est une Unité Mixte de Recherche (UMR 7162) du Centre National de la Recherche Scientifique (CNRS) et de l’Université Paris Cité.
Actualités

Sara Ducci élue Optica Fellow
Toutes nos félicitations à Sara Ducci, Professeure au Laboratoire Matériaux et Phénomènes Quantiques (MPQ, Université Paris Cité / CNRS), qui vient d'être élue Fellow de Optica (anciennement OSA), "pour ses réalisations significatives dans le domaine de la photonique...
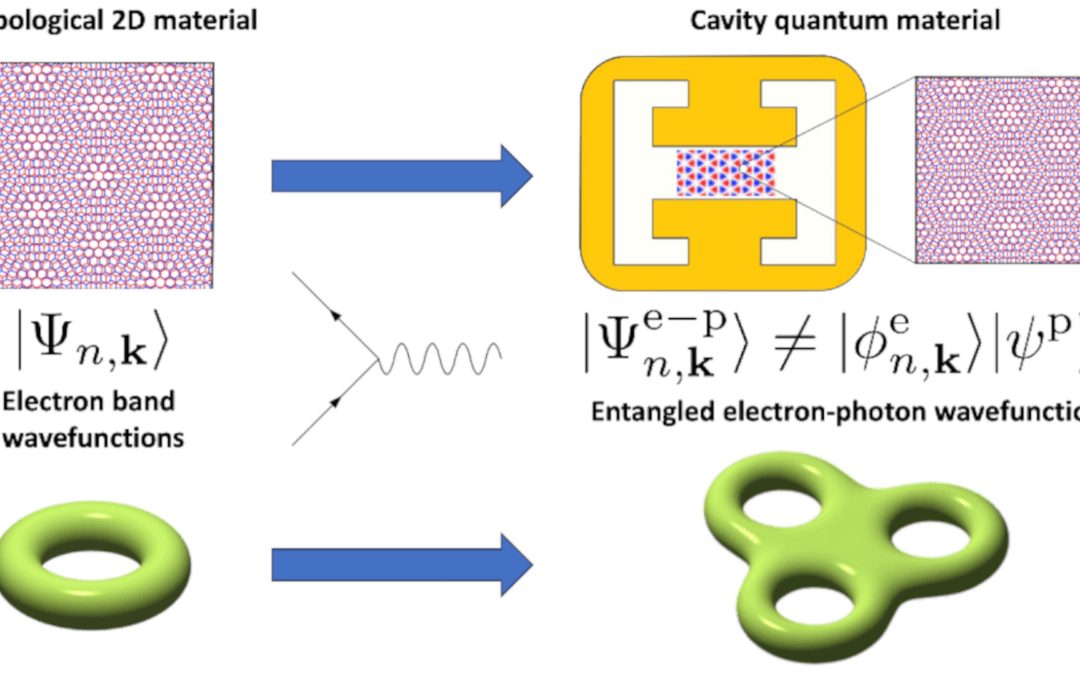
Nouvelle topologie des états électron-photon dans les matériaux quantiques en cavité
L'équipe THEORIE vient de publier un article dans Phys. Rev. Lett. sur les propriétés tologogiques des matériaux quantiques en cavité. Les matériaux topologiques présentent des propriétés extraordinaires, grâce aux caractéristiques uniques de leurs fonctions d'onde...

Atelier “A la découverte des technologies quantiques” – Fête de la science
A l'occasion de la fête de la science 2023, un atelier sur les technologiques quantiques est organisé par le laboratoire MPQ (équipe QITE) en partenariat avec l'IRIF, les 9 et 10 octobre : conférence sur la cryptographie, atelier sur les photons intriqués et sur les...

Hommage à Jean Klein
Nous avons eu la tristesse d’apprendre le décès le 29 juillet 2023 de Jean Klein, professeur à l’UFR de Physique depuis sa création. Ingénieur de Supélec (promo 1962), Jean Klein débute sa carrière de chercheur au Groupe de Physique des Solides de l’ENS sous la...
 Dispositifs Optiques Nonlinéaires (DON)
Dispositifs Optiques Nonlinéaires (DON)
 Quantum Information and Technologies (QITE)
Quantum Information and Technologies (QITE)
 Equipe THEORIE
Equipe THEORIE
 Microscopie Electronique Avancée et Nano-Structures (MEANS)
Microscopie Electronique Avancée et Nano-Structures (MEANS)
 Auto-organisation de nanostructures et STM
Auto-organisation de nanostructures et STM
 Spectroscopie de QUAsi-Particules (SQUAP)
Spectroscopie de QUAsi-Particules (SQUAP)
 Transport Electronique à L’Echelle Moléculaire (TELEM)
Transport Electronique à L’Echelle Moléculaire (TELEM)
Tutelles

